
Points clés
- L’essai ne présente pas de biais méthodologique. Les résultats sont cohérents et non discutables. Il s’agit d’une véritable innovation thérapeutique et percée scientifique !
- L’effet clinique observé après 18 mois est, au mieux, modeste
- Le caractère « disease-modifier » de ce médicament, permettant de penser que le bénéfice du médicament va s’accentuer avec le temps, reste à l’heure actuelle contestable
- Le risque d’ARIA grave est plutôt rare mais pas rarissime : attention en pratique courante !
- Le coût médico-économique s’annonce probablement très élevé
- Il apparaît important de bien anticiper et discuter des conditions d’utilisation de ces médicaments en cas d’AMM devant leur balance bénéfice/risque discutable pour le moment
Un essai méthodologique propre, une innovation thérapeutique et « percée » pour la science !
Les résultats de la phase 3 du lecanemab constituent les données de la plus haute qualité et de la plus grande cohérence qui aient été divulguées et publiées à ce jour sur les immunothérapies anti-amyloïdes aux stades précoces de la maladie d’Alzheimer (MA) [1]. Les résultats et la méthodologie sont univoques et ne peuvent être contestés. Eisai a révélé des détails supplémentaires lors de la présentation au congrès CTAD (Clinical Trials on Alzheimer’s Disease, San Francisco, 2022) qui ont renforcé les statistiques et le message de base de l’article du New England Journal of Medicine. Nous ne pouvons qu’être satisfaits après le désastre de la saga aducanumab ! Thibaud Lebouvier, Julien Delrieu, Olivier Hanon, et Maria Soto les ont très bien détaillés dans l’article « Historique ! », nous ne les redétaillerons pas ici.
En tant que scientifiques, nous ne pouvons que nous réjouir de ces résultats sans ambiguïté qui représentent une véritable percée dans la thérapeutique de la MA : l’utilisation d’anticorps monoclonaux dirigés contre un peptide censé jouer un rôle dans la physiopathologie de la MA a un effet clinique démontré dans la maladie !
En tant que médecins, nous avons des sentiments plus mitigés quant au rapport bénéfice/risque de ce médicament.
Des bénéfices cliniques statistiquement significatifs mais, au mieux, modestes
En ce qui concerne les bénéfices, après 18 mois, nous observons une diminution de 27% du déclin cognitif mesuré sur la CDR-SB (Clinical Dementia Rating scale–Sum of Boxes), soit 0,45 points. Cette différence de 0,45 points sur le déclin cognitif mesuré entre les bras placebo et traités est tout à fait cohérente avec ce qui avait été observé dans les récents résultats des essais thérapeutiques impliquant des immunothérapies anti-amyloïdes forte dose testées aux stades précoces de la MA : l’essai de phase 2 du lecanemab (0,40 points après 18 mois), la phase 2 du donanemab (0,36 points après 18 mois), les essais de phase 3 du gantenerumab forte dose (0,31 et 0,19 points après 27 mois dont 18 mois de forte dose) et un des deux essais de phase 3 de l’aducanumab (essai EMERGE : 0,39 points après 18 mois) [2,3]. Cela s’écarte en revanche des résultats de l’essai de phase 3 négatif de l’aducanumab (essai ENGAGE – 0,03 points après 18 mois).
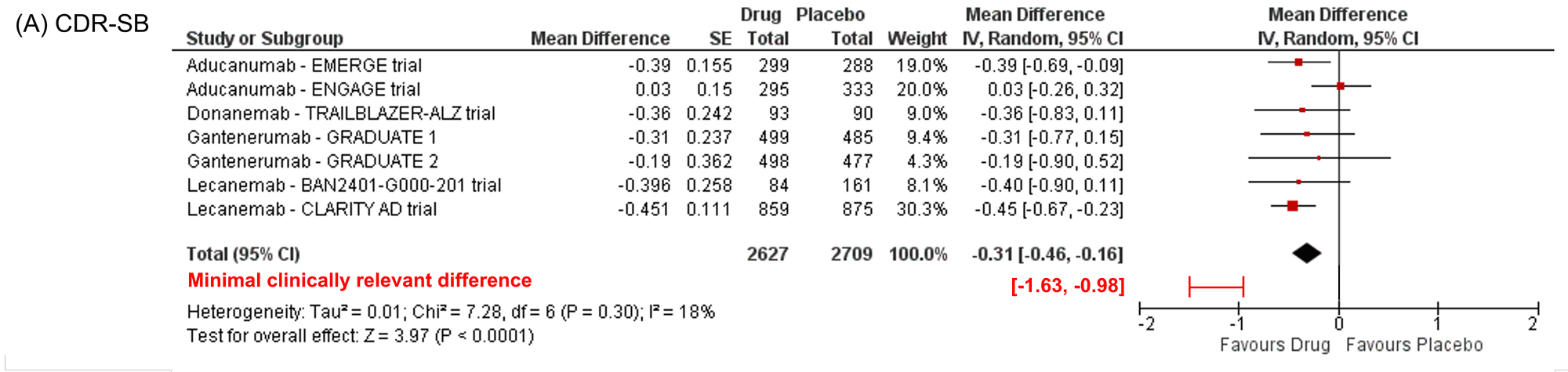
La CDR-SB est une échelle peu couramment utilisée en clinique mais qui fait consensus dans de nombreux essais cliniques aux stades précoces de la maladie d’Alzheimer. Il s’agit d’une échelle hybride sur 18 points (0 étant l’absence de trouble, 18 le stade le plus avancé d’un trouble neurocognitif majeur) évaluant les fonctions cognitives et les capacités fonctionnelles du sujet, avec une pondération particulière pour les troubles mnésiques et leurs conséquences. L’échelle CDR permet de calculer deux scores : le score global, qui permet une classification en 5 stades d’un trouble neurocognitif (CDR 0 : absence de trouble ; CDR 0,5 : trouble neurocognitif léger/incertain ; CDR 1 : trouble neurocognitif majeur à un stade léger ; CDR 2 : trouble neurocognitif majeur à un stade modéré ; CDR 3 : trouble neurocognitif majeur à un stade sévère), et la « somme des boites » qui rend un score de 0 à 18 avec des demi points. Les équivalences entre le score global et la SB ont été établies comme suit : CDR 0 : CDR-SB = 0 ; CDR 0,5 : CDR-SB = 0,5-4 ; CDR 1 : CDR-SB = 4,5-9 ; CDR 2 : CDR-SB = 9,5-15,5 ; CDR 3 : CDR-SB = 16-18 [4]. Dans l’essai de phase 3 du lecanemab, les patients avaient en moyenne à l’inclusion 3,17 (groupe traité) et 3,22 (groupe placebo) points de CDR-SB. Le groupe placebo a atteint en moyenne 4,88 points de CDR-SB à la fin des 18 mois de l’étude et le groupe traité 4,38 points. En 2019, des chercheurs du groupe Eli Lilly avaient cherché à déterminer quelles étaient les valeurs minimales cliniquement pertinentes sur l’échelle CDR-SB pour un traitement donné (c’est-à-dire les valeurs à partir desquelles un médecin spécialiste est capable de dire s’il y a eu, ou non, évolution clinique même minimale). Au terme d’un très beau travail fait sur plus de 20 000 participants de la cohorte académique américaine NACC (National Alzheimer’s Coordinating Center) avec un suivi longitudinal et en utilisant plusieurs méthodologies, ils avaient retenu que ces valeurs minimales correspondaient à une différence de 1 point sur l’échelle CDR-SB pour les patients au stade de trouble neurocognitif léger et de 2 points pour les patients au stade de ‘démence légère’ [5]. Bien que, par définition, la pertinence clinique est une donnée subjective, ces mêmes valeurs ont été retrouvées par des chercheurs de Roche sur 769 patients avec un trouble cognitif léger de type amnésique suivis pendant 12 mois lors d’un essai clinique de phase 3 (essai ADC-008 comparant l’efficacité de la vitamine E vs. donepezil et conduit entre 1999 et 2004) [6]. Les valeurs communiquées par Eisai et Biogen concernant l’efficacité du lecanemab semblent donc bien en deçà de ces valeurs minimales cliniquement pertinentes. Les effets sur la qualité de vie sont bien sûr encourageants et importants, mais les récentes revues de la littérature concernant ces échelles remettent en cause leur pertinence dans les essais cliniques sur la MA [7].
Un argument souvent entendu est qu’il s’agit de traitements dits « disease-modifiers » : le bénéfice observé à 18 mois n’est donc qu’un début et continuera de s’amplifier avec le temps pour atteindre ces valeurs cliniquement pertinentes. Il y a des arguments pour et contre concernant cette assertion (biologiques, effets non cohérents sur tous les biomarqueurs, courbes d’évolution qui se parallélisent après 12 mois…) et ceci n’est pas formellement démontré à ce jour. Aussi, si la différence absolue de 0,45 point sur la CDR-SB, et non la différence relative de 27 %, était la différence maximale réelle pouvant être atteinte avec le lecanemab, cela limiterait sa pertinence clinique. Pour renforcer le niveau de preuve d’un effet « disease-modifier » du médicament, de nouveaux essais cliniques type « delayed-start » pourraient permettre de mieux estimer l’efficacité attendue à long terme.
Attention aux ARIAs graves : plutôt rares mais pas rarissimes !
Néanmoins, en tant que cliniciens nous avons aussi l’habitude de nous contenter de « petits gains » notamment si les risques et la gêne liés au traitement sont minimes ou négligeables (la fameuse balance bénéfice/risque). Qu’en est-il donc des risques ? Le risque majeur de cette classe de médicaments est les ARIA (Amyloid –Related Imaging Abnormalities) : des réactions oedémateuses et/ou hémorragiques cérébrales qui s’apparentent aux formes sporadiques inflammatoires d’angiopathie amyloïde. Le lecanemab n’y échappe pas : 21,3% des patients traités ont développé une ARIA pendant le suivi (contre 9,3% dans le groupe placebo). Nous avons l’habitude de dire et lire que ces ARIA sont bénignes, et nécessitent simplement une courte suspension de traitement [8]. De fait, un seul cas d’ARIA grave sous aducanumab forte dose a été rapporté jusqu’ici dans la littérature [9]. Nous venons malheureusement d’en rapporter deux autres issus de cet essai de phase 3 du lecanemab (avec malheureusement des séquelles à long terme) [2], et 3 décès possiblement imputables au lecanemab, dont un semble-t-il non discutable, se sont produits ces 6 derniers mois aux Etats-Unis durant la phase d’extension [10]. Concernant les 18 mois de l’essai randomisé : trois cas graves d’ARIA-E (severe ARIA-E) ont été rapportés lors de la présentation au CTAD. En outre, 10/898 (1,1 %) des patients du groupe lecanemab étaient classés comme ayant des effets indésirables graves avec ARIA (serious adverse event with ARIA), contre 1/897 (0,1%) dans le groupe placebo (Dr Michael Irizarry, communication personnelle). Deux de ces cas sont survenus à la Pitié-Salpêtrière, et nous les avons décrits dans un article publié en septembre dernier (Fig 3. de [2]). La patiente avec l’ARIA-H grave décrite dans cet article a été revue fin novembre 2022 : elle a dû être institutionnalisée, elle est quasi-mutique avec une apraxie de la marche, son MMSE est désormais de 4/30, 18 mois après l’ARIA (25/30 un mois avant l’ARIA) sans qu’un autre événement intercurrent n’explique cette rapide dégradation. Ces ARIA graves peuvent être associées à des séquelles à long terme et représentent une perte de chance pour le patient. Leur taux réel reste inconnu mais ces chiffres provisoires semblent indiquer que le risque d’ARIA grave est plutôt rare mais pas non plus rarissime (ce d’autant plus qu’il est ici estimé à partir de données issues d’essais thérapeutiques où les critères d’exclusion sont rigoureusement respectés, le suivi très rapproché, et les protocoles de gestion des ARIAs suivis à la lettre ; le risque sera peut-être accru en pratique courante). Le risque d’ARIA grave peut être exacerbé, comme la fréquence des ARIA, par l’utilisation de fortes doses d’immunothérapies anti-amyloïdes, ce qui est la tendance actuelle. Ce résultat nous fait faire un parallèle lointain avec l’AN1792, cette immunothérapie active (=vaccin) anti-amyloïde interrompue dans sa phase 2a en 2003 du fait de méningo-encéphalites graves survenant chez 6% des patients [11]. A posteriori, les aspects d’imagerie de ces méningo-encéphalites ressemblent beaucoup aux ARIA graves que nous décrivons actuellement [12]. Les données de sécurité du lecanemab nous appellent donc à la vigilance et doivent être mises en balance avec ce ralentissement de 0,45 points sur la CDR-SB après 18 mois de traitement. Par ailleurs, il s’agit d’une perfusion IV bi-mensuelle dans cet essai avec des surveillances IRM régulières, les contraintes ne sont donc pas nulles pour les patients (même si tous les industriels travaillent actuellement à des formulations sous-cutanées avec des dispositifs d’auto-injection qui réduiront un peu la lourdeur du protocole de soin).
Analyses en sous-groupes
Globalement, le rapport bénéfice/risque de ce médicament peut être remis en question, malgré la qualité méthodologique de l’étude. Il apparaît donc crucial d’identifier au travers d’analyses post-hoc les profils de patients présentant un potentiel meilleur rapport bénéfice/risque de ces traitements. De ce point de vue, les analyses post-hoc (comme toujours à interpréter avec prudence, même si le statut APOE était en l’occurrence un stratum de randomisation, limitant les biais de ces analyses post-hocs) montrent une absence d’effet clinique chez les patients porteurs homozygotes de l’allèle ε4 de l’APOE. Les patients non porteurs semblent quant à eux non seulement tirer un bénéfice plus important du lecanemab mais aussi avoir moins de risque de développer des ARIAs.
Challenge médico-économique
En tant que médecins de CMRR, n’appartenant pas aux agences gouvernementales d’évaluation du médicament, nous ne considérons pas que notre rôle est d’émettre un avis médico-économique sur ces médicaments. C’est pour cela que nous nous attardons ici surtout sur la balance bénéfice/risque individuelle. Ce sont néanmoins des dimensions qui seront soulevées par nos autorités notamment au moment des négociations sur le remboursement. Sans crier au loup, les immunothérapies restent des traitements coûteux à produire pour les industriels (il suffit de regarder le coût de 1661,12€ d’une dose de 1000mg de rituximab désormais génériqué, dose pour un adulte dans le cadre d’une utilisation hors AMM chez un patient souffrant de SEP), et il y aura de nombreux coûts indirects associés (plus fort recours aux biomarqueurs, augmentation de l’activité médicale et paramédicale pour le diagnostic et la surveillance, surveillance IRM des ARIAs, …). Pour les agences du médicament et les payeurs, la décision s’annonce difficile compte tenu de la prévalence élevée de la MA (nous avons estimé à environ 300 000 le nombre de patients potentiellement éligibles à ces traitements en France [13]), du coût total prévisionnel élevé et de la complexité de mise en œuvre du lecanemab dans le système de soin.
Conclusions et perspectives
D’autres données issues de médicaments de la même classe vont arriver prochainement : le donanemab d’Eli Lilly (mi-2023). Ceci nous permettra de mieux estimer cette balance bénéfice/risque.
Il faut aussi garder en tête que les grandes avancées thérapeutiques dans l’histoire de la médecine ne se sont pas toujours accompagnées de molécules pionnières au profil bénéfice/risque important (par exemple l’AZT en monothérapie pour le VIH, la tacrine dans la MA, …) mais qu’elles sont le déclencheur du développement ultérieur de nouveaux médicaments ou de polythérapies ayant un meilleur rapport bénéfice/risque.
La récente intervention du PDG d’Eisai nous rend optimistes [14]. Il démontre la volonté d’Eisai de prendre en compte ces préoccupations lorsqu’il affirme que “ce n’est pas un remède” et préconise une prescription prudente chez les homozygotes APOE4. L’identification – et peut-être, dans un premier temps, la limitation – des indications à des sous-groupes présentant un meilleur rapport bénéfice/risque, et une meilleure démonstration de l’effet « disease-modifier » dans des nouveaux essais cliniques (de type « delayed-start »), peuvent être des solutions pour mieux aligner les préoccupations des patients, de l’industriel, des prescripteurs, des agences de régulation du médicament et des payeurs.
Nous serons heureux de contribuer à ces débats et discussions au sein des groupes de travail qui seront prochainement mis en place par la FCM. Cela peut être un grand pas pour le domaine et les patients : faisons les choses bien !
Dr Nicolas Villain, Neurologue, CMRR Paris Sud (Pitié-Salpêtrière)
Dr Vincent Planche, Neurologue, CMRR de Bordeaux
Dr Stéphanie Bombois, Neurologue, CMRR Paris Sud (Pitié-Salpêtrière)
Références
[3] Bateman R, Smith J, Donohue M, Delmar P, Abbas R, Salloway S, et al. TOPLINE RESULTS OF PHASE III GRADUATE I & II PIVOTAL TRIALS WITH SUBCUTANEOUS GANTENERUMAB. 16TH Clin. TRIALS ALZHEIMER’S Dis., San Francisco: 2022.
[14] How will new Alzheimer’s drug lecanemab be prescribed? n.d. (accessed December 23, 2022).
